
+91 6002993949
submission@iarconsortium.org
Open Access
ISSN (Print) : 2788-8843
ISSN (Online) : 2788-8851
Type 2 Diabetes mellitus (T2DM) is characterized by high blood sugar caused by a lack of insulin, insulin resistance, or both. It's linked to the onset of secondary problems, which can lead to a variety of co-morbidities. Recent research has found that diabetics are more likely to acquire cognitive impairment or dementia. Diabetes is linked to a number of neurological illnesses, including Alzheimer's disease (AD). Evidence of a relationship between diabetes and AD is growing. Insulin signalling disruption in the brain has been discovered, resulting in increased tau protein phosphorylation (hyperphosphorylation), a hallmark and diagnostic of AD pathology, and the buildup of neurofibrillary tangles (NFT). Insulin malfunction in the brain has been shown to modify glycogen synthase kinase-3β (GSK-3β) activity, resulting in increased β amyloid and tau phosphorylation in diabetics. GSK-3β signalling has been implicated in the physiological and pathological processes of diabetes and AD, respectively. This could explain why diabetic individuals have a higher chance of developing AD as their diabetes progresses and they get older. Interestingly, several in vivo investigations with oral antidiabetic medications and insulin treatment in diabetic patients showed improved cognitive function and lower tau hyperphosphorylation. The relationship between T2DM and AD as it relates to amyloid and tau pathology will be discussed in this article. A better knowledge of the relationship between T2DM and AD could transform how researchers and doctors handle both diseases in the future, potentially leading to new therapies and prevention techniques.
Type 2 Diabetes mellitus (T2DM) is characterized by high blood sugar caused by a lack of insulin, insulin resistance, or both. It's linked to the onset of secondary problems, which can lead to a variety of co-morbidities. Recent research has found that diabetics are more likely to acquire cognitive impairment or dementia. Diabetes is linked to a number of neurological illnesses, including Alzheimer's disease (AD) [1,2,3,4,5]. The most prevalent cause of dementia in the elderly is AD. Extracellular β amyloid (Aβ) containing senile plaques, intracellular hyperphosphorylated Tau containing neurofibrillary tangles (NFT), neuroinflammation, synapse loss, and neuronal death are all symptoms of AD. Aging and the allele of the apolipoprotein E 4 are both known risk factors. T2DM has recently been identified as an additional risk factor for AD. T2DM and AD have comparable pathophysiologies, such as insulin resistance, altered glucose and lipid metabolism, inflammation, and oxidative stress, according to mounting data [1,2,3,4,5].
Once upon a time, the brain was supposed to be an insulin-insensitive organ. Insulin, on the other hand, is now universally acknowledged to play a significant role in neuronal survival and brain function. Insulin is essential for brain synaptic plasticity, which aids learning and memory [6]. Insulin also promotes the production of dendritic spines and synapse, neural stem cell activation, neurite growth and repair, and neuroprotection [7]. As a result, changes in insulin metabolism and signalling in the Central Nervous System (CNS) can play a role in the development of a variety of mental illnesses. Many animal and clinical investigations have demonstrated a link between neurodegenerative illnesses like AD and altered insulin signalling in the central nervous system over the last 25 years [8,9]; B, demonstrating that insulin resistance and decreased insulin activity may play a role in the aetiology of certain brain diseases through several pathways. Following that, we'll go over the primary pathophysiological links between AD and T2DM, emphasising the role of a malfunctioning insulin transduction pathway in neurodegenerative determinism. We'll look at how insulin signalling and Glycogen synthase kinase 3β (GSK3β) play a role in the creation of intracellular neurofibrillary tangles (NFTs) and the deposition of Aβ plaques, two hallmarks of AD pathology.
Molecular Mechanism of Insulin Resistant in type 2 Diabetes Mellitus with Alzheimer's Disease
Insulin resistance in the brain is becoming better recognised as a component in the development of AD. A robust link between insulin signalling and Aβ metabolism has been discovered in several investigations. Aβ oligomers, such as dimers, trimers, and dodecamers (Aβ *56), are particularly harmful in AD. GSK3β is activated by cerebral insulin resistance, resulting in an increase in Aβ synthesis and Tau phosphorylation [10,11,12]. Insulin resistance was found to increase extracellular Aβ deposition by increasing the activity of the enzyme γ - secretase, which is involved in Aβ synthesis, and therefore encouraging Aβ secretion from neurons [13]. Insulin, on the other hand, inhibits GSK3β activity, preventing the formation of Aβ and hyperphosphorylated Tau [14]. The transgenic animals displayed hippocampus insulin resistance, according to studies employing transgenic AD mouse models [15]. After being fed a high fat diet, leptin deficient mice, a T2DM model, showed altered brain insulin signaling and cognitive deficits [16,17]. Insulin receptor substrate-1 (IRS-1) phosphorylation at serine residues, a hallmark of insulin resistance, was shown to be significantly increased in postmortem AD brains. Insulin resistance is also linked to a reduction in synaptic plasticity [18]. In an AD animal model, insulin treatment reduced chronic neuroinflammation and microglia activation while also improving synapse formation [19,2]. These investigations demonstrated links between AD, cerebral insulin resistance, and T2DM.
Figure 1 depicts a model that links between T2DM, cerebral insulin resistance, and AD pathogenesis. Through GSK3β, hyperinsulinemia, oxidative stress, and advanced glycation end products (AGEs), T2DM and the metabolic syndrome may exacerbate AD pathogenesis [20] Hyperinsulinemia in T2DM may impair A clearance through competitive inhibition of the insulin degrading enzyme (IDE), which is a key regulator of Aβ levels in neural cells. Through a number of shared or contemporaneous pathways involving predisposing genes and environmental variables, brain insulin resistance may develop in tandem with T2DM. T2DM related hyperinsulinemia may cause brain insulin resistance by reducing insulin receptor expression and receptor kinase activity [21] and, as a result, increasing Aβ and tau pathology. Inversely, or even reciprocally, abnormal oligomeric or fibrillar Aβ can cause brain insulin resistance. Aβ has a similar sequence to insulin and can attach to the insulin receptor directly, causing insulin resistance.
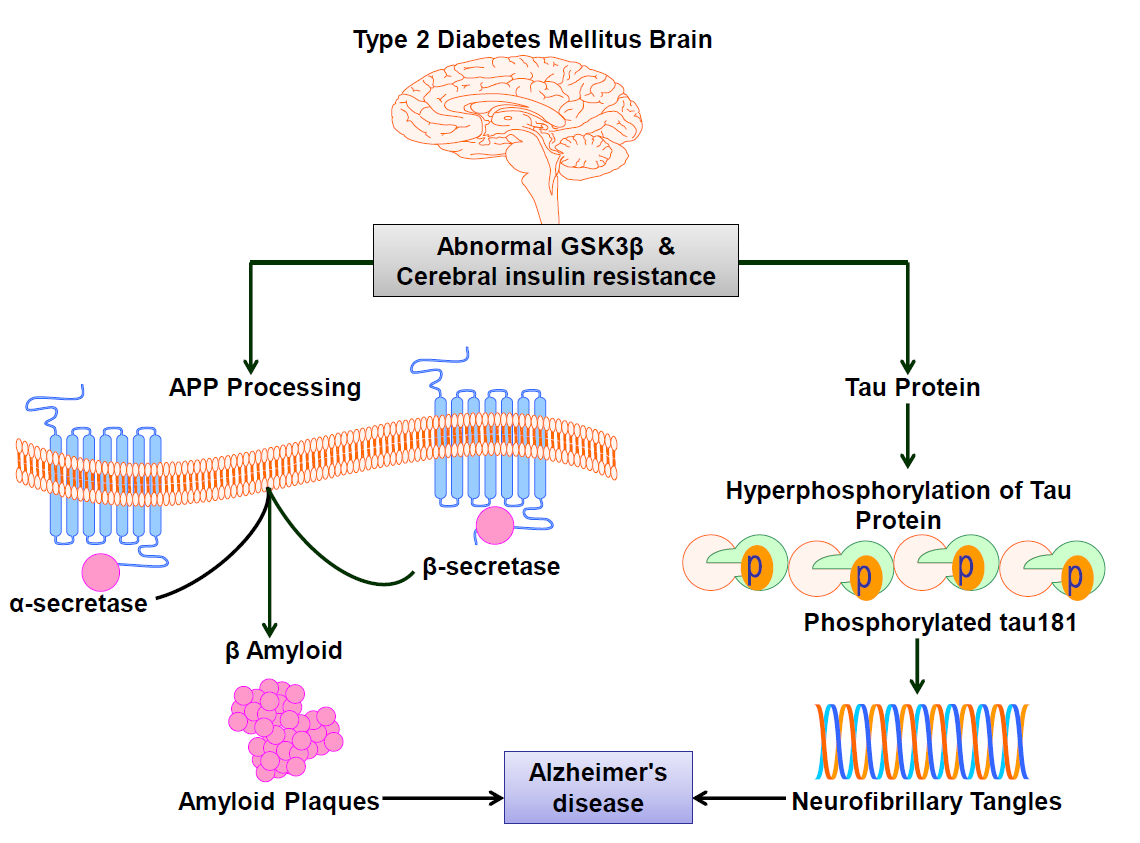
Figure 1: Type 2 Diabetes Mellitus Brain
Molecular Mechanism of GSK3β in type 2 Diabetes Mellitus with Alzheimer's Disease
GSK3β is a key regulatory kinase that plays a role in a variety of processes including glycogen metabolism, apoptosis, protein synthesis, cell signalling, cell transport, gene transcription, proliferation, and intracellular communication. Many substances linked to AD have been discovered to interact with it, including the microtubule-associated protein tau, presenilin 1, the Aβ peptide, amyloid precursor protein (APP), and acetylcholine. GSK3β may also have a role in brain ageing and lifespan [22].
T2DM is caused by a lack of insulin secretion caused by islet β cell failure, which can be congenital or acquired. Although the specific mechanism is unknown, it could be linked to glucose toxicity, lipid toxicity, inflammatory response, oxidative stress, and other variables [23,24,25]. GSK3β is one of the primary mediators of islet β cell apoptosis, and it's linked to insulin insufficiency [25]. Excessive activation of GSK3β resulted in a reduction in islet β cell proliferation in DM model mice [26]. Islet β cells are endocrine cells in the body that release insulin, which helps to regulate blood sugar levels. Endogenous GSK3β inhibits the PI3K/Akt signaling pathway, which controls islet β cell development, and therefore plays a key role in blood glucose regulation.
Insulin modulates the equilibrium between Aβ anabolism and catabolism via regulating peripheral Aβ and tau metabolism, which controls Aβ release in the brain through modulating APP metabolism [27]. T2DM and AD may be linked by changes in Aβ production and degradation caused by insulin deficiency or action. Deficiencies in insulin dependent pathways may increase GSK3β activation, which has been linked to an increased risk of AD. In the context of Aβ toxicity, T2DM alters mitochondrial antioxidant mechanisms and supports brain weakening [27].
A previous clinical study found that hyperglycemia induced islet β cell loss is associated with increased oxidative stress, and that some enzymatic markers of oxidative stress are similar in mild cognitive impairment (MCI) and AD patients, implying that oxidative damage could be a key factor in the development of severe cognitive impairment [28]. According to studies, elevated oxidative stress causes APP cleavage and Aβ generation, and increased Aβ causes LPO levels [29,30]. According to Grimes and Jope [31], oxidative stress activates GSK3β in neuronal cells, while GSK3β inhibition regulates oxidative stress in neuronal hippocampus cell lines [32]. GSK3β and oxidative stress are linked, according to this discovery.
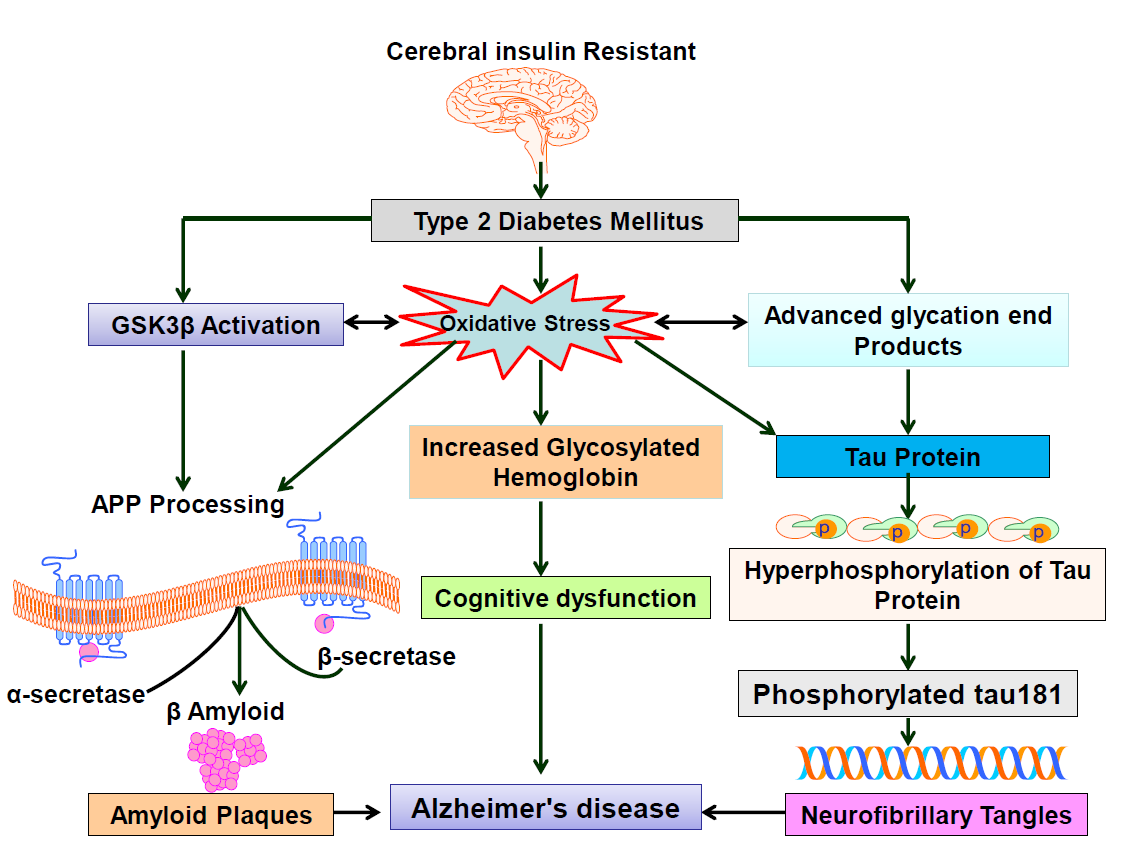
Figure 2: Cerebral insulin Resistant
We endeavoured to highlight the rising body of literature that portrays the shared pathophysiology of T2DM and AD, as well as expound on the underlying molecular pathways at the crossroads of these two diseases, in this review. GSK3β is the main rate limiting enzyme for glycogen production suppression in T2DM. More significantly, it is one of the leading causes of insulin insufficiency and resistance, and insulin resistance is a hallmark of T2DM development and progression. GSK3β is involved in both hyperphosphorylation (NFT creation) and APP metabolism (Aβ generation) in AD. While insulin resistance in T2DM can generate Aβ deposition, which is removed by tau, excessive tau phosphorylation can exacerbate Aβ neurotoxicity, damage the brain, and impair cognitive function. GSK3β may not only be a promising therapeutic target, but also a key hint to overcoming the AD Mountain. T2DM with AD has the potential to deliver a multitude of preventative and therapeutic techniques to existing patients, despite the fact that the specific mechanisms connecting T2DM and AD remain convoluted and ambiguous, which may have disastrous socioeconomic repercussions on public health and healthcare systems. For the time being, it appears that more anti T2DM medications with positive benefits against cognitive impairment will be investigated.
Talbot, K. et al. “Demonstrated Brain Insulin Resistance in Alzheimer’s Disease Patients Is Associated with IGF-1 Resistance, IRS-1 Dysregulation, and Cognitive Decline.” The Journal of Clinical Investigation, vol. 122, no. 4, 2012, pp. 1316–1338.
Chen, Y. et al. “Deregulation of Brain Insulin Signaling in Alzheimer’s Disease.” Neuroscience Bulletin, vol. 30, no. 2, 2014, pp. 282–294.
Yaffe, K. et al. “Diabetes, Impaired Fasting Glucose, and Development of Cognitive Impairment in Older Women.” Neurology, vol. 63, no. 4, 2004, pp. 658–663.
Mushtaq, G. et al. “Biological Mechanisms Linking Alzheimer’s Disease and Type-2 Diabetes Mellitus.” CNS & Neurological Disorders—Drug Targets, vol. 13, no. 7, 2014, pp. 1192–1201.
Willette, A.A. et al. “Association of Insulin Resistance with Cerebral Glucose Uptake in Late Middle-Aged Adults at Risk for Alzheimer Disease.” JAMA Neurology, vol. 72, no. 9, 2015, pp. 1013–1020.
Chiu, S.L. et al. “Insulin Receptor Signaling Regulates Synapse Number, Dendritic Plasticity, and Circuit Function in Vivo.” Neuron, vol. 58, no. 5, 2008, pp. 708–719.
Apostolatos, A. et al. “Insulin Promotes Neuronal Survival via the Alternatively Spliced Protein Kinase CδII Isoform.” Journal of Biological Chemistry, vol. 287, no. 12, 2012, pp. 9299–9310.
Kleinridders, A. et al. “Insulin Action in Brain Regulates Systemic Metabolism and Brain Function.” Diabetes, vol. 63, no. 7, 2014, pp. 2232–2243.
Biessels, G.J. et al. “Dementia and Cognitive Decline in Type 2 Diabetes and Prediabetic Stages: Towards Targeted Interventions.” The Lancet Diabetes & Endocrinology, vol. 2, no. 3, 2014, pp. 246–255.
De Felice, F.G. “Alzheimer’s Disease and Insulin Resistance: Translating Basic Science into Clinical Applications.” The Journal of Clinical Investigation, vol. 123, no. 2, 2013, pp. 531–539.
Avrahami, L. et al. “Inhibition of Glycogen Synthase Kinase-3 Ameliorates β-Amyloid Pathology and Restores Lysosomal Acidification and Mammalian Target of Rapamycin Activity in the Alzheimer Disease Mouse Model.” Journal of Biological Chemistry, vol. 288, no. 2, 2013, pp. 1295–1306.
Phiel, C.J. et al. “GSK-3α Regulates Production of Alzheimer’s Disease Amyloid-β Peptides.” Nature, vol. 423, no. 6938, 2003, pp. 435–439.
Son, S.M. et al. “Altered APP Processing in Insulin-Resistant Conditions Is Mediated by Autophagosome Accumulation via the Inhibition of Mammalian Target of Rapamycin Pathway.” Diabetes, vol. 61, no. 12, 2012, pp. 3126–3138.
DaRocha-Souto, B. et al. “Activation of Glycogen Synthase Kinase-3 Beta Mediates β-Amyloid–Induced Neuritic Damage in Alzheimer’s Disease.” Neurobiology of Disease, vol. 45, no. 1, 2012, pp. 425–437.
Bomfim, T.R. et al. “An Anti-Diabetes Agent Protects the Mouse Brain from Defective Insulin Signaling Caused by Alzheimer’s Disease–Associated Aβ Oligomers.” The Journal of Clinical Investigation, vol. 122, no. 4, 2012, pp. 1339–1353.
Gao, C. et al. “New Animal Models of Alzheimer’s Disease That Display Insulin Desensitization in the Brain.” Reviews in the Neurosciences, vol. 24, no. 6, 2013, pp. 607–615.
Ramos-Rodriguez, J.J. et al. “Differential Central Pathology and Cognitive Impairment in Pre-Diabetic and Diabetic Mice.” Psychoneuroendocrinology, vol. 38, 2013, pp. 2462–2475.
Grillo, C. et al. “Hippocampal Insulin Resistance Impairs Spatial Learning and Synaptic Plasticity.” Diabetes, vol. 64, 2015, pp. 3927–3936.
Adzovic, L. et al. “Insulin Improves Memory and Reduces Chronic Neuroinflammation in the Hippocampus of Young but Not Aged Brains.” Journal of Neuroinflammation, vol. 12, no. 1, 2015, pp. 1–10.
Eldar-Finkelman, H., and Krebs, E.G. “Phosphorylation of Insulin Receptor Substrate 1 by Glycogen Synthase Kinase 3 Impairs Insulin Action.” Proceedings of the National Academy of Sciences, vol. 94, no. 18, 1997, pp. 9660–9664.
Kim, B. et al. “Cortical Neurons Develop Insulin Resistance and Blunted Akt Signaling: A Potential Mechanism Contributing to Enhanced Ischemic Injury in Diabetes.” Antioxidants & Redox Signaling, vol. 14, no. 10, 2011, pp. 1829–1839.
Vinothkumar, G. et al. “Glycogen Synthase Kinase 3β: A Key Player of Cognitive Dysfunction in Chronic Kidney Disease Patients and a Possible Link between Abnormal pTau and Platelet APP Processing and Therapeutic Role of Erythropoietin.” Personalized Medicine in Psychiatry, vol. 25, 2021, p. 100073.
Nolan, C.J. “Lipotoxicity, β Cell Dysfunction, and Gestational Diabetes.” Cell Metabolism, vol. 19, no. 4, 2014, pp. 553–554.
Donath, M.Y. et al. “Inflammatory Mediators and Islet β-Cell Failure: A Link between Type 1 and Type 2 Diabetes.” Journal of Molecular Medicine, vol. 81, no. 8, 2003, pp. 455–470.
Robertson, R.P. et al. “Chronic Oxidative Stress as a Mechanism for Glucose Toxicity of the Beta Cell in Type 2 Diabetes.” Cell Biochemistry and Biophysics, vol. 48, no. 2, 2007, pp. 139–146.
Liu, Z. et al. “Mice with Beta Cell Overexpression of Glycogen Synthase Kinase-3β Have Reduced Beta Cell Mass and Proliferation.” Diabetologia, vol. 51, no. 4, 2008, pp. 623–631.
Suzanne, M. “Contributions of Brain Insulin Resistance and Deficiency in Amyloid-Related Neurodegeneration in Alzheimer’s Disease.” Drugs, vol. 72, no. 1, 2012, pp. 49–66.
Padurariu, M. et al. “Changes of Some Oxidative Stress Markers in the Serum of Patients with Mild Cognitive Impairment and Alzheimer’s Disease.” Neuroscience Letters, vol. 469, no. 1, 2010, pp. 6–10.
Butterfield, D.A. “β-Amyloid-Associated Free Radical Oxidative Stress and Neurotoxicity: Implications for Alzheimer’s Disease.” Chemical Research in Toxicology, vol. 10, no. 5, 1997, pp. 495–506.
Tabner, B.J. et al. “Hydrogen Peroxide Is Generated during the Very Early Stages of Aggregation of the Amyloid Peptides Implicated in Alzheimer Disease and Familial British Dementia.” Journal of Biological Chemistry, vol. 280, no. 43, 2005, pp. 35789–35792.
Grimes, C.A., and Jope, R.S. “The Multifaceted Roles of Glycogen Synthase Kinase 3β in Cellular Signaling.” Progress in Neurobiology, vol. 65, no. 4, 2001, pp. 391–426.
Lee, K.Y. et al. “Glycogen Synthase Kinase-3β Activity Plays Very Important Roles in Determining the Fate of Oxidative Stress-Inflicted Neuronal Cells.” Brain Research, vol. 1129, 2007, pp. 89–99.
